scVIVA for representing cells and their environment in spatial transcriptomics#
In this tutorial, we go through the steps of training scVIVA, a deep generative model that leverages both cell-intrinsic and neighboring gene expression profiles to produce stochastic embeddings of cell states as well as normalized gene expression profiles. We show how to obtain informative fine-grained partitions of cells that reflects both their internal state and the surrounding tissue and use the generative model to test hypotheses of differential expression between tissue niches.
Plan for this tutorial:
Loading the data
Training a scVIVA model
Visualizing the latent space
Perform DE analysis across niches
%load_ext autoreload
%autoreload 2
# Install from GitHub for now
!pip install --quiet spatialvi-tools
!pip install --quiet adjustText
import os
import random
import tempfile
import numpy as np # type: ignore
import scanpy as sc # type: ignore
import spatialvi # type: ignore
import torch # type: ignore
from rich import print # type: ignore
sc.set_figure_params(figsize=(4, 4))
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={'facecolor' : "w"}
%config InlineBackend.figure_format='retina'
spatialvi.settings.seed = 0
random.seed(42)
np.random.seed(42)
torch.manual_seed(42)
print("Last run with spatialvi-tools version:", spatialvi.__version__)
Seed set to 0
Last run with spatialvi-tools version: 0.1.0
# Quickly check the correct folder is used.
spatialvi.__file__
'/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/spatialvi/__init__.py'
Data loading#
In this tutorial we load a human breast cancer section, generated with 10X Xenium. The cell segmentation originally performed on this data resulted in many erroneously assigned transcripts and therefore re-segmented the cells using the ProSeg algorithm, which is a scalable algorithm for transcriptome-informed segmentation.
adata_path = os.path.join(save_dir.name, "adata_for_tuto_s1.h5ad")
adata = sc.read(
adata_path,
backup_url="https://exampledata.scverse.org/scvi-tools/adata_for_tuto_s1.h5ad",
)
adata
AnnData object with n_obs × n_vars = 117305 × 313
obs: 'sample', 'x', 'y', 'z', 'observed_x', 'observed_y', 'observed_z', 'fov', 'n_counts', 'index', 'cell_type'
var: 'mt'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'log1p', 'pca'
obsm: 'X_pca', 'X_scANVI', 'spatial'
varm: 'PCs'
layers: 'counts', 'counts_log1p', 'counts_wo_bg', 'min_max_scaled', 'min_max_scaled_raw'
The authors identified distinct tumor domains in this specimen, corresponding to in situ ductal carcinoma (DCIS) and invasive tumor:
sc.pl.spatial(adata, color="cell_type", spot_size=30)
/tmp/ipykernel_2295422/2186789331.py:1: FutureWarning: Use `squidpy.pl.spatial_scatter` instead.
sc.pl.spatial(adata, color="cell_type", spot_size=30)
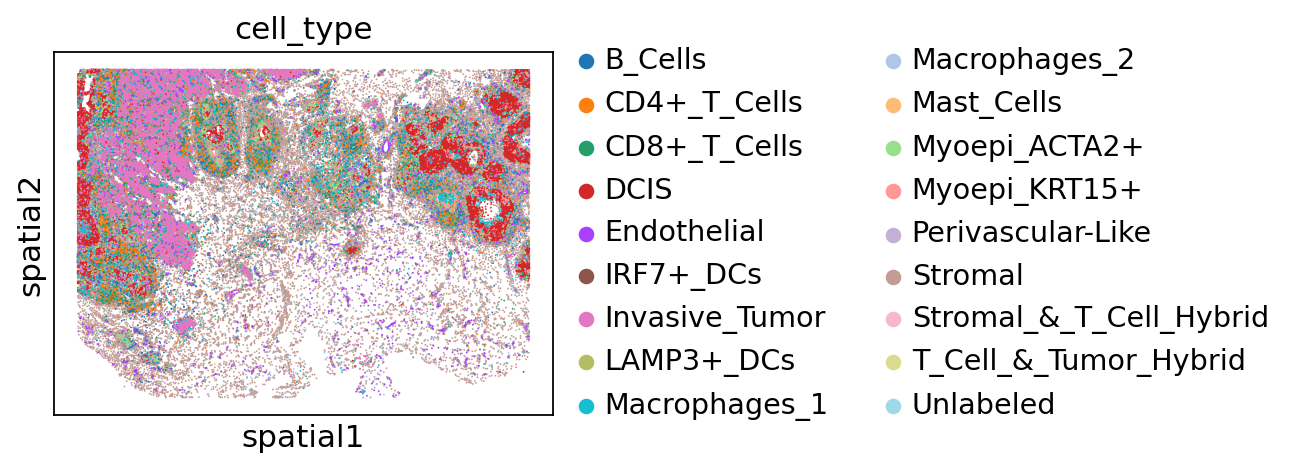
adata.obs["cell_type"].value_counts()
cell_type
Stromal 32426
Invasive_Tumor 18185
DCIS 15993
CD4+_T_Cells 9387
Macrophages_1 8644
CD8+_T_Cells 7493
Endothelial 7000
B_Cells 5359
Myoepi_ACTA2+ 5324
Myoepi_KRT15+ 2636
Macrophages_2 2616
Perivascular-Like 817
Unlabeled 516
IRF7+_DCs 454
LAMP3+_DCs 218
Mast_Cells 203
Stromal_&_T_Cell_Hybrid 28
T_Cell_&_Tumor_Hybrid 6
Name: count, dtype: int64
Train scVIVA model#
We first define the neighborhood of each cell using a k-nn graph. We set \(k=20\). Then, the environment features are defined in two ways - the first is the cell-type composition of its cellular neighborhood. The second is the average gene expression state of neighboring cells, with a separate profile for each of the present cell types. These cell-intrinsic gene expression states can be learned with a spatially unaware model such as scANVI, or with resolVI.
Here we assume that scANVI has already been trained on the data, and that the embeddings are stored in the AnnData object. We refer to the scANVI tutorial for training the model.
Environment features computations occur in the preprocessing_anndata method, that adds the relevant keys to the AnnData object.
setup_kwargs = {
"sample_key": "sample", # column in adata.obs that contains the individual slide ID
"labels_key": "cell_type", # column in adata.obs that contains the cell type labels
"cell_coordinates_key": "spatial", # spatial coordinates key in adata.obsm
"expression_embedding_key": "X_scANVI", # expression embedding key in adata.obsm
}
from spatialvi.model._scviva import SCVIVA
spatialvi.model.SCVIVA.preprocessing_anndata(
adata,
k_nn=20, # number of nearest neighbors for spatial graph construction
**setup_kwargs,
)
Saved niche_indexes and niche_distances in adata.obsm
Saved niche_composition in adata.obsm
Saved niche_activation in adata.obsm
Then, as in all scvi-tools model, we need to register the AnnData.
spatialvi.model.SCVIVA.setup_anndata(
adata,
layer="counts", # adata layer that contains the raw counts
batch_key="sample", # column in adata.obs that contains the batch covariate
**setup_kwargs,
)
INFO Using column names from columns of adata.obsm['niche_composition']
INFO Generating sequential column names
INFO Generating sequential column names
INFO Generating sequential column names
INFO Generating sequential column names
INFO Generating sequential column names
We instantiate a scVIVA model:
nichevae = spatialvi.model.SCVIVA(adata)
nichevae
scVIVA model with the following parameters: n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: poisson, latent_distribution: normal. Training status: Not Trained Model's adata is minified?: False
nichevae.train(
max_epochs=600,
early_stopping=True,
check_val_every_n_epoch=1,
batch_size=512,
plan_kwargs={
"lr": 5e-4,
},
)
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
💡 Tip: For seamless cloud logging and experiment tracking, try installing [litlogger](https://pypi.org/project/litlogger/) to enable LitLogger, which logs metrics and artifacts automatically to the Lightning Experiments platform.
You are using a CUDA device ('NVIDIA RTX 6000 Ada Generation') that has Tensor Cores. To properly utilize them, you should set `torch.set_float32_matmul_precision('medium' | 'high')` which will trade-off precision for performance. For more details, read https://pytorch.org/docs/stable/generated/torch.set_float32_matmul_precision.html#torch.set_float32_matmul_precision
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/lightning/pytorch/utilities/_pytree.py:21: `isinstance(treespec, LeafSpec)` is deprecated, use `isinstance(treespec, TreeSpec) and treespec.is_leaf()` instead.
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/lightning/pytorch/trainer/connectors/data_connector.py:434: The 'train_dataloader' does not have many workers which may be a bottleneck. Consider increasing the value of the `num_workers` argument` to `num_workers=63` in the `DataLoader` to improve performance.
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/lightning/pytorch/trainer/connectors/data_connector.py:434: The 'val_dataloader' does not have many workers which may be a bottleneck. Consider increasing the value of the `num_workers` argument` to `num_workers=63` in the `DataLoader` to improve performance.
Monitored metric elbo_validation did not improve in the last 45 records. Best score: 237.645. Signaling Trainer to stop.
We can plot the training curves:
nichevae.history.keys()
dict_keys(['kl_weight', 'validation_loss', 'elbo_validation', 'reconstruction_loss_validation', 'kl_local_validation', 'kl_global_validation', 'niche_compo_validation', 'niche_reconst_validation', 'train_loss', 'elbo_train', 'reconstruction_loss_train', 'kl_local_train', 'kl_global_train', 'niche_compo_train', 'niche_reconst_train'])
Let’s plot for instance the validation ELBO, niche composition and state losses:
nichevae.history["elbo_validation"].plot()
<Axes: xlabel='epoch'>
nichevae.history["niche_compo_validation"].plot()
<Axes: xlabel='epoch'>
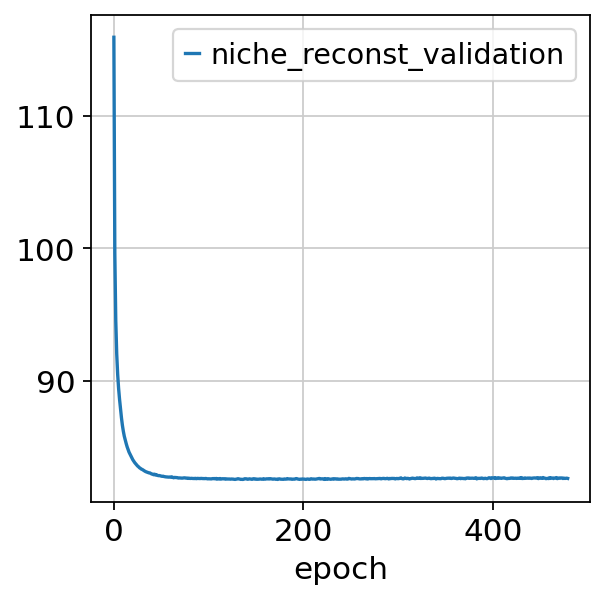
nichevae.history["niche_reconst_validation"].plot()
<Axes: xlabel='epoch'>
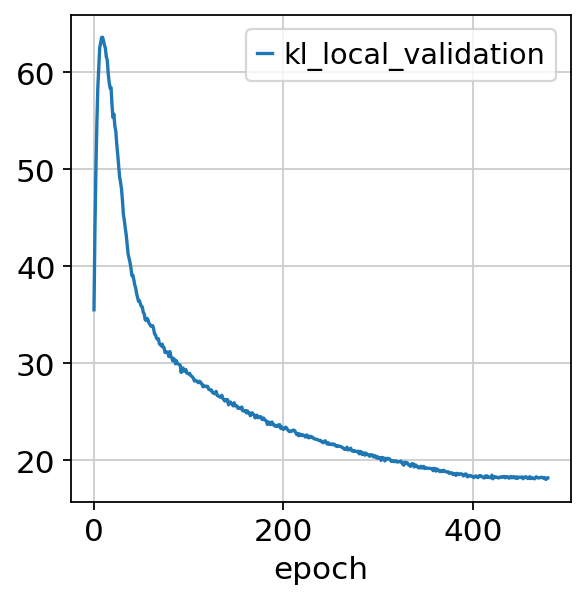
nichevae.history["kl_local_validation"].plot()
<Axes: xlabel='epoch'>
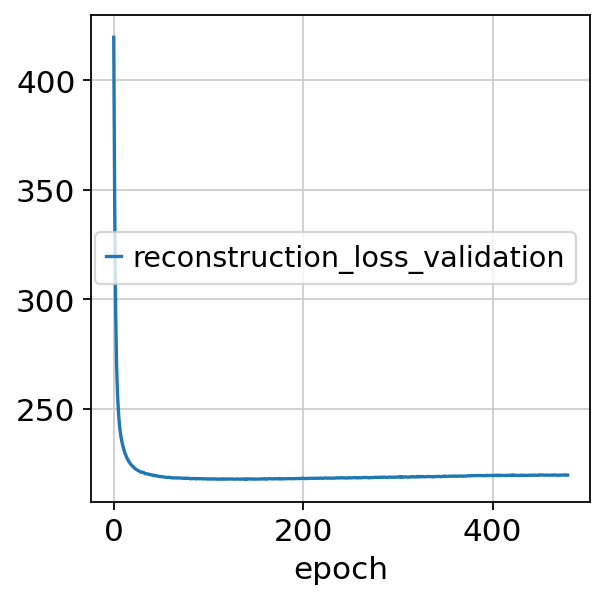
nichevae.history["reconstruction_loss_validation"].plot()
<Axes: xlabel='epoch'>
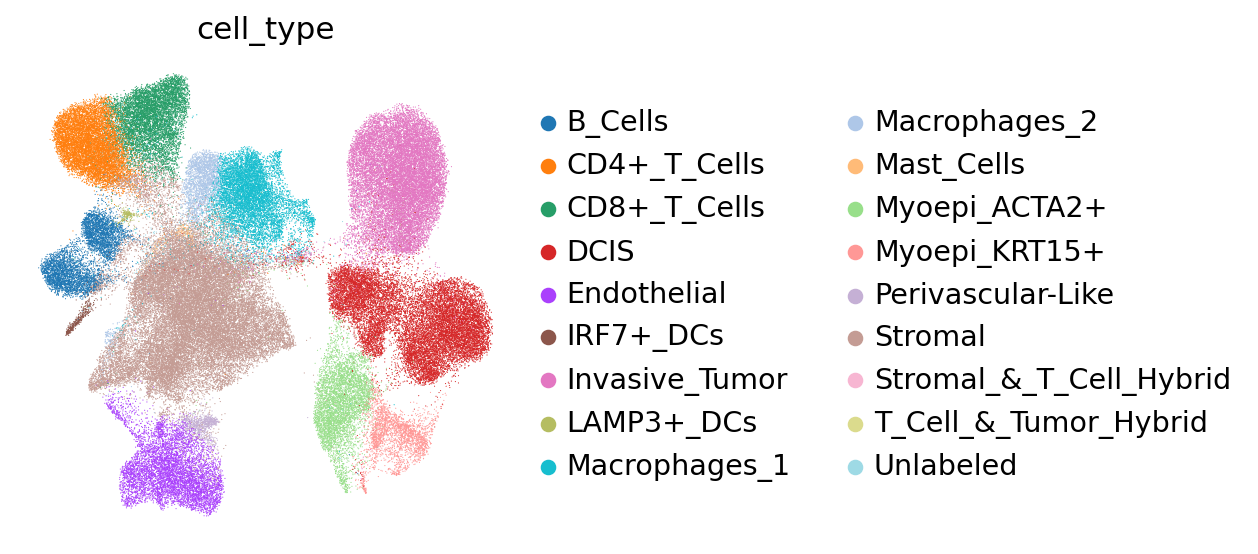
After training the model, we can compute and store the latent space:
adata.obsm["X_scVIVA"] = nichevae.get_latent_representation()
We may visualize the latent space in UMAP coordinates, coloring by cell type.
sc.pp.neighbors(adata, use_rep="X_scVIVA", n_neighbors=30)
sc.tl.umap(adata)
sc.pl.umap(adata, color="cell_type", frameon=False)
Differential expression analysis#
We now use the generative model to test hypotheses of differential expression between the niches. We’ll focus on endothelial cells.
adata_endothelial = adata[adata.obs["cell_type"] == "Endothelial"].copy()
adata_not_endo = adata[adata.obs["cell_type"] != "Endothelial"].copy()
print(adata_endothelial)
AnnData object with n_obs × n_vars = 7000 × 313 obs: 'sample', 'x', 'y', 'z', 'observed_x', 'observed_y', 'observed_z', 'fov', 'n_counts', 'index', 'cell_type', '_scvi_batch', '_scvi_labels', '_scvi_sample' var: 'mt' uns: '_scvi_manager_uuid', '_scvi_uuid', 'log1p', 'pca', 'cell_type_colors', 'cell_type_to_int', 'neighbors', 'umap' obsm: 'X_pca', 'X_scANVI', 'spatial', 'niche_indexes', 'niche_distances', 'niche_composition', 'niche_activation', 'X_scVIVA', 'X_umap' varm: 'PCs' layers: 'counts', 'counts_log1p', 'counts_wo_bg', 'min_max_scaled', 'min_max_scaled_raw' obsp: 'distances', 'connectivities'
We perform coarse Leiden clustering on the endothelial latent space, in a bid to find spatially confined populations of endothelial cells.
sc.pp.neighbors(adata_endothelial, use_rep="X_scVIVA", n_neighbors=30, random_state=42)
sc.tl.umap(adata_endothelial)
sc.tl.leiden(
adata_endothelial,
key_added="leiden_scVIVA",
resolution=0.3,
flavor="igraph",
n_iterations=-1,
random_state=42,
)
adata_endothelial.obs["leiden_scVIVA"].unique() # check the number of clusters
['0', '1', '2', '3', '4']
Categories (5, object): ['0', '1', '2', '3', '4']
We focus on clusters 0 and 1, which are located in the stromal and tumor regions, respectively. We then perform differential expression analysis between these two clusters.
sc.pl.spatial(
adata_endothelial,
color="leiden_scVIVA",
spot_size=80,
groups=["1", "0"],
)
/tmp/ipykernel_2295422/3415536345.py:1: FutureWarning: Use `squidpy.pl.spatial_scatter` instead.
sc.pl.spatial(

adata.obs["leiden_scVIVA"] = "Unknown"
adata.obs.loc[adata.obs["cell_type"] == "Endothelial", "leiden_scVIVA"] = adata_endothelial.obs[
"leiden_scVIVA"
]
adata.obs["leiden_scVIVA"].value_counts()
leiden_scVIVA
Unknown 110305
1 2410
4 1997
3 1745
0 564
2 284
Name: count, dtype: int64
We now run the differential expression function, between the cell groups \(\textit{G1}=tumor~endothelial\) and \(\textit{G2}=stromal~endothelial\). We first set the number of nearest neighbors to compute the non-endothelial neighbors of \(\textit{G1}\) and \(\textit{G2}\), called \(\textit{N1}\) and \(\textit{N2}\), respectively.
Setting niche_mode=True, we compute 4 different DE tests: \(\{\textit{G1}~vs~\textit{G2}\}\), \(\{\textit{G1}~vs~\textit{N1}\}\), \(\{\textit{N1}~vs~\textit{G2}\}\) and \(\{\textit{N1}~vs~\textit{N2}\}\) (in this order). We set a test-specific treshold for significant log-fold change DELTA.
Other parameters include the number of samples to draw from the posterior N_SAMPLES_DE, PSEUDOCOUNTS for stability and FDR for the FDR correction. More details can be found in Boyeau et al. PNAS 2023.
delta_niches = 0.05 # smaller delta for niche comparison
delta_markers = 0.15 # bigger delta for G1-N1 comparison
DELTA = [delta_niches, delta_markers, delta_niches, delta_niches]
K_NN_DE = 6
GROUP = "leiden_scVIVA"
G1 = "1"
G2 = "0"
PSEUDOCOUNTS = 1e-4
N_SAMPLES_DE = 1e5
FDR = 0.2
DE_1_0 = nichevae.differential_expression(
adata,
groupby=GROUP,
group1=G1,
group2=G2,
k_nn=K_NN_DE,
delta=DELTA,
niche_mode=True,
n_samples_overall=N_SAMPLES_DE,
fdr_target=FDR,
pseudocounts=PSEUDOCOUNTS,
)
Computing adjusted nearest neighbors...
Using 1 samples
Mean number of neighbors: 3.3 ± 2.1
Computing DE...
Running DE for group1_group2
Running DE for group1_neighbors1
Running DE for neighbors1_group2
Running DE for neighbors1_neighbors2
Computing g1 confidence scores...
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/sklearn/gaussian_process/kernels.py:450: ConvergenceWarning: The optimal value found for dimension 0 of parameter k1__constant_value is close to the specified upper bound 1000.0. Increasing the bound and calling fit again may find a better value.
warnings.warn(
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/sklearn/gaussian_process/kernels.py:450: ConvergenceWarning: The optimal value found for dimension 0 of parameter k2__alpha is close to the specified upper bound 1.0. Increasing the bound and calling fit again may find a better value.
warnings.warn(
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/sklearn/gaussian_process/kernels.py:440: ConvergenceWarning: The optimal value found for dimension 0 of parameter k2__length_scale is close to the specified lower bound 10.0. Decreasing the bound and calling fit again may find a better value.
warnings.warn(
Let’s analysize the DE test: \(\textit{G1}=tumor~endothelial\) vs \(\textit{G2}=stromal~endothelial\). The DE function returns a Dataclass object DE_1_0.
We can access the Gaussian process classifier properties with the gpc attribute:
DE_1_0.gpc
GaussianProcessClassifier(kernel=1**2 * RationalQuadratic(alpha=0.1, length_scale=10),
n_restarts_optimizer=10, random_state=0)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
DE_1_0.gpc_info()
Training score: 0.9032258064516129
Marginal likelihood: -29.282496339215044
Kernel: 31.6**2 * RationalQuadratic(alpha=1, length_scale=10)
The \(\textit{G1}\) vs \(\textit{G2}\) differential expression results are stored in the g1_g2 attribute:
DE_1_0.g1_g2
| proba_de | proba_not_de | bayes_factor | scale1 | scale2 | pseudocounts | delta | lfc_mean | lfc_median | lfc_std | ... | raw_mean2 | non_zeros_proportion1 | non_zeros_proportion2 | raw_normalized_mean1 | raw_normalized_mean2 | is_de_fdr_0.2 | comparison | group1 | group2 | proba_de_g1_n1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADH1B | 0.99907 | 0.00093 | 6.979385 | 0.000325 | 0.042892 | 0.0001 | 0.05 | -7.355366 | -7.399907 | 2.398989 | ... | 13.189716 | 0.040249 | 0.695035 | 3.004496 | 361.016815 | True | 1 vs 0 | 1 | 0 | 0.000000 |
| VOPP1 | 0.99322 | 0.00678 | 4.986974 | 0.006631 | 0.002813 | 0.0001 | 0.05 | 1.314478 | 1.309808 | 0.563384 | ... | 0.962766 | 0.616598 | 0.510638 | 66.419924 | 32.232405 | True | 1 vs 0 | 1 | 0 | 0.123607 |
| ESM1 | 0.99182 | 0.00818 | 4.797848 | 0.010812 | 0.000169 | 0.0001 | 0.05 | 5.423343 | 5.505044 | 2.151102 | ... | 0.063830 | 0.429876 | 0.046099 | 119.344614 | 2.532916 | True | 1 vs 0 | 1 | 0 | 0.959440 |
| TCIM | 0.98829 | 0.01171 | 4.435532 | 0.020674 | 0.005132 | 0.0001 | 0.05 | 2.028386 | 2.051936 | 0.833136 | ... | 1.888298 | 0.743568 | 0.556738 | 208.053761 | 59.586497 | True | 1 vs 0 | 1 | 0 | 0.977427 |
| ADIPOQ | 0.98783 | 0.01217 | 4.396536 | 0.000083 | 0.011207 | 0.0001 | 0.05 | -6.656808 | -6.853134 | 2.796564 | ... | 3.592199 | 0.009959 | 0.434397 | 0.589076 | 98.371829 | True | 1 vs 0 | 1 | 0 | 0.000000 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| CD27 | 0.47566 | 0.52434 | -0.097437 | 0.000069 | 0.000069 | 0.0001 | 0.05 | -0.083514 | -0.089351 | 1.479412 | ... | 0.015957 | 0.011618 | 0.015957 | 0.603928 | 0.390254 | False | 1 vs 0 | 1 | 0 | 0.000000 |
| APOBEC3A | 0.47558 | 0.52442 | -0.097758 | 0.000047 | 0.000041 | 0.0001 | 0.05 | 0.138008 | 0.128841 | 0.845916 | ... | 0.015957 | 0.006639 | 0.010638 | 0.442661 | 0.795928 | False | 1 vs 0 | 1 | 0 | 0.007082 |
| KLRC1 | 0.45763 | 0.54237 | -0.169887 | 0.000144 | 0.000143 | 0.0001 | 0.05 | -0.006569 | 0.005273 | 0.876546 | ... | 0.056738 | 0.017427 | 0.037234 | 1.019192 | 1.906989 | False | 1 vs 0 | 1 | 0 | 0.124016 |
| TCL1A | 0.44339 | 0.55661 | -0.227415 | 0.000030 | 0.000040 | 0.0001 | 0.05 | -0.144663 | -0.109740 | 1.310926 | ... | 0.017730 | 0.009129 | 0.014184 | 0.569848 | 0.693454 | False | 1 vs 0 | 1 | 0 | 0.000000 |
| CCL20 | 0.42274 | 0.57726 | -0.311535 | 0.000038 | 0.000034 | 0.0001 | 0.05 | 0.040833 | 0.026631 | 1.046383 | ... | 0.015957 | 0.003320 | 0.014184 | 0.190507 | 0.418955 | False | 1 vs 0 | 1 | 0 | 0.012565 |
313 rows × 23 columns
Where the probability of true DE according to the Gaussian process classifier is stored in the proba_de_g1_n1 column:
DE_1_0.g1_g2["proba_de_g1_n1"]
ADH1B 0.000000
VOPP1 0.123607
ESM1 0.959440
TCIM 0.977427
ADIPOQ 0.000000
...
CD27 0.000000
APOBEC3A 0.007082
KLRC1 0.124016
TCL1A 0.000000
CCL20 0.012565
Name: proba_de_g1_n1, Length: 313, dtype: float64
We may also access the other tests results in the same way: DE_1_0.g1_n1, DE_1_0.n1_g2 and DE_1_0.n1_n2.
We can then filter genes to upregulated genes, i.e. such that the median Log-Fold Change over the samples is positive, and the proba_de (ratio of LFC greater than the defined delta treshold over the total number of posterior samples) is greater than a given filter - here we set it to 0.8.
PROBA_TRES = 0.8
g1_g3_genes = DE_1_0.g1_g2[
(DE_1_0.g1_g2["lfc_median"] > 0) & (DE_1_0.g1_g2["proba_de"] > PROBA_TRES)
].index
We then display the results: median Log-Fold Change (LFC) of upregulated genes in \(\textit{G1}\) vs \(\textit{G2}\) displayed on the x-axis, while we compare differential expression computed between \(\textit{N1}\) and \(\textit{G2}\) on the y-axis.
Genes are colored by their marker label (yellow=significantly upregulated in \(\textit{G1}\) vs \(\textit{N1}\), green otherwise).
We also display the classifier decision boundary (the predicted probability of being in the yellow class).
PLOT_MARGIN = 0.2
DE_1_0.plot(
filter=g1_g3_genes, # selected genes to plot
# path_to_save="DE_plot.svg",
margin=PLOT_MARGIN, # margin around the plot
legend_loc="upper right", # location of the legend
)
/opt/anaconda3/envs/scvi_new/lib/python3.13/site-packages/sklearn/utils/validation.py:2691: UserWarning: X does not have valid feature names, but GaussianProcessClassifier was fitted with feature names
warnings.warn(
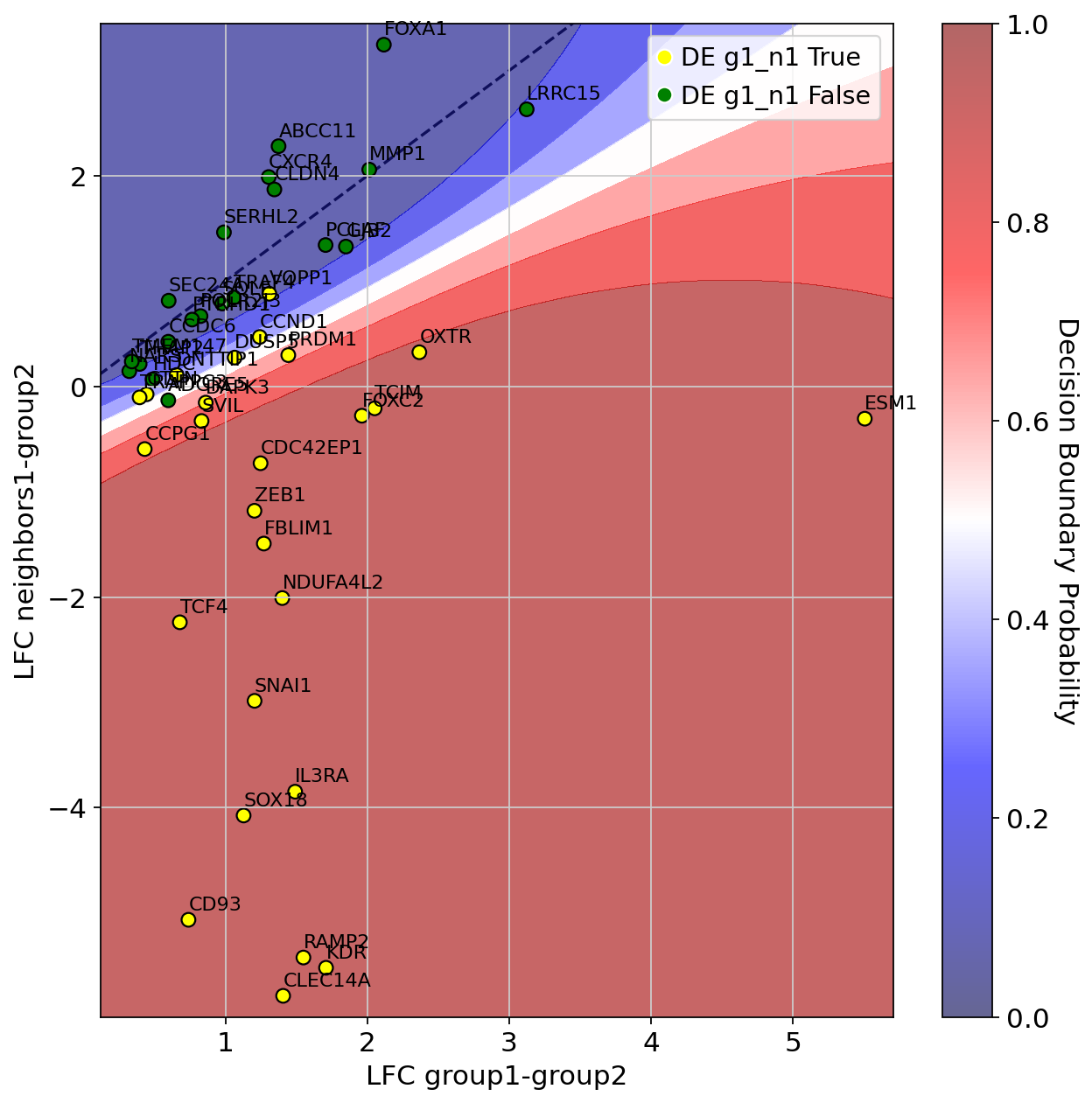
You can select the marker genes (positive class for the classifier, yellow in the plot):
DE_1_0.gpc.confident_genes
Index(['VOPP1', 'ESM1', 'TCIM', 'ZEB1', 'CDC42EP1', 'PRDM1', 'DNTTIP1',
'DAPK3', 'CCND1', 'KDR', 'RAMP2', 'OXTR', 'FOXC2', 'NDUFA4L2',
'CLEC14A', 'FBLIM1', 'DUSP5', 'IL3RA', 'CTTN', 'SOX18', 'SVIL', 'SNAI1',
'CCPG1', 'TCF4', 'TRAPPC3', 'CD93'],
dtype='object')
We can also check the predicted class probabilities of the Gaussian process classifier:
DE_1_0.g1_g2["proba_de_g1_n1"].loc[DE_1_0.gpc.confident_genes]
VOPP1 0.123607
ESM1 0.959440
TCIM 0.977427
ZEB1 0.995345
CDC42EP1 0.984172
PRDM1 0.687767
DNTTIP1 0.343943
DAPK3 0.749674
CCND1 0.382805
KDR 0.984881
RAMP2 0.985878
OXTR 0.928817
FOXC2 0.979331
NDUFA4L2 0.998993
CLEC14A 0.980917
FBLIM1 0.997867
DUSP5 0.459823
IL3RA 0.996980
CTTN 0.390382
SOX18 0.995834
SVIL 0.851219
SNAI1 0.998668
CCPG1 0.854724
TCF4 0.998267
TRAPPC3 0.387794
CD93 0.987385
Name: proba_de_g1_n1, dtype: float64
Then we can further filter the confident gene list, by setting a treshold on the classifier predictions - for instance 0.9:
DE_1_0.g1_g2["proba_de_g1_n1"].loc[DE_1_0.gpc.confident_genes][
DE_1_0.g1_g2["proba_de_g1_n1"].loc[DE_1_0.gpc.confident_genes] > 0.9
].index
Index(['ESM1', 'TCIM', 'ZEB1', 'CDC42EP1', 'KDR', 'RAMP2', 'OXTR', 'FOXC2',
'NDUFA4L2', 'CLEC14A', 'FBLIM1', 'IL3RA', 'SOX18', 'SNAI1', 'TCF4',
'CD93'],
dtype='object')
Finally, we can plot spatial maps of the selected genes. We first compute global percentiles of the gene expression values to set an upper bound for the color scale.
def get_gene_percentiles_list(adata, gene_list, p, layer=None):
"""
Calculate the p-percentile of gene expression for a list of genes in an AnnData object.
Parameters
----------
adata (AnnData): The AnnData object containing expression data.
gene_list (list): List of gene names for which to compute percentiles.
p (float): Percentile to compute (between 0 and 100).
layer (str or None): The layer from which to retrieve expression data.
If None, uses `adata.X`.
Returns
-------
list: A list of p-percentile values for the genes, in the same order as gene_list.
If a gene is not found, its value will be `None`.
"""
percentiles = []
for gene in gene_list:
if gene in adata.var_names:
if layer:
data = adata[:, gene].layers[layer].flatten()
else:
data = adata[:, gene].X.flatten()
# Compute the percentile
percentiles.append(np.percentile(data, p))
else:
percentiles.append(None) # Handle genes not in adata.var_names
return percentiles
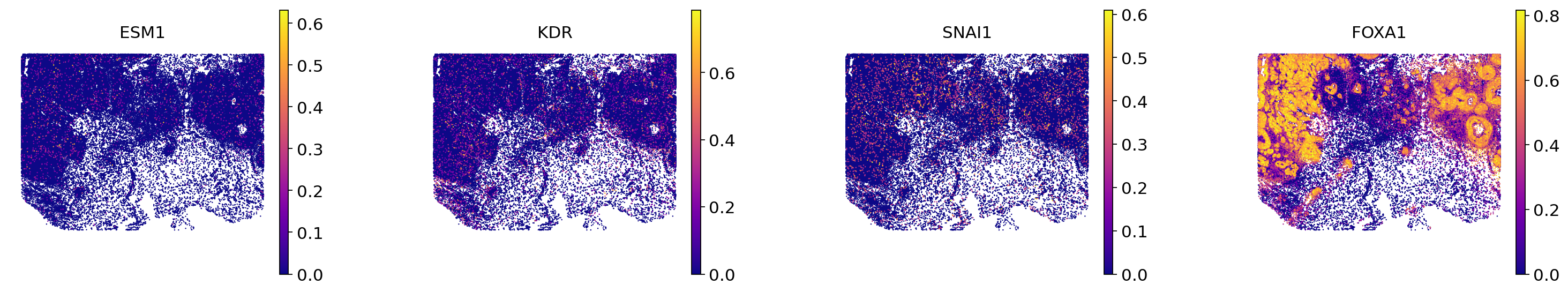
We display ESM1, KDR, SNAI1, critical genes for angiogenesis in invasive cancer. We aslo display FOXA1, that is both upregulated in \(\textit{G1}\) and \(\textit{N1}\), to show how our procedure can filter such genes.
gene_list_invasive = ["ESM1", "KDR", "SNAI1", "FOXA1"]
percentiles_invasive = get_gene_percentiles_list(
adata, gene_list_invasive, 99.9, layer="min_max_scaled"
)
We first plot these genes in endothelial cells:
plot_endo = True
sc.pl.spatial(
adata=adata_endothelial if plot_endo else adata_not_endo,
spot_size=100 if plot_endo else 40,
color=gene_list_invasive,
frameon=False,
use_raw=False,
wspace=0.4,
vmax=percentiles_invasive,
layer="min_max_scaled",
cmap="plasma",
)
/tmp/ipykernel_2295422/3718530031.py:3: FutureWarning: Use `squidpy.pl.spatial_scatter` instead.
sc.pl.spatial(
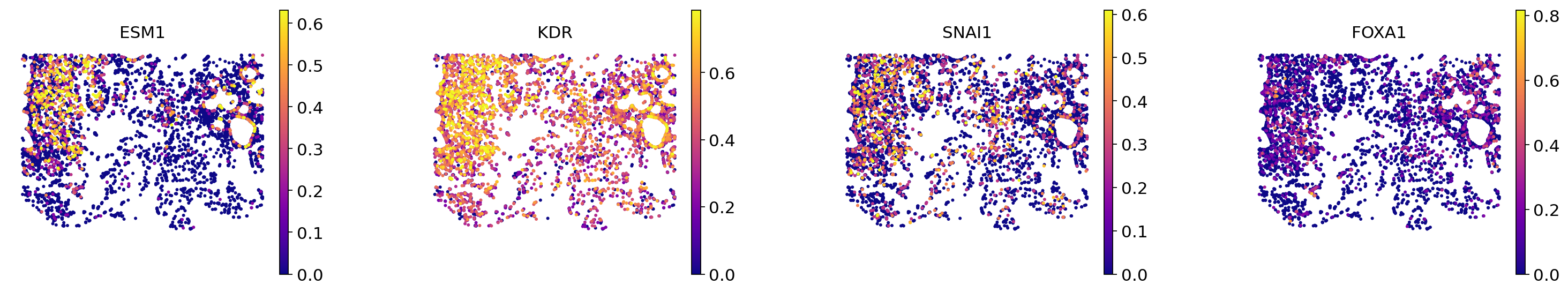
Then in all cells but endothelial:
plot_endo = False
sc.pl.spatial(
adata=adata_endothelial if plot_endo else adata_not_endo,
spot_size=100 if plot_endo else 40,
color=gene_list_invasive,
frameon=False,
use_raw=False,
wspace=0.4,
vmax=percentiles_invasive,
layer="min_max_scaled",
cmap="plasma",
)
/tmp/ipykernel_2295422/1505343501.py:3: FutureWarning: Use `squidpy.pl.spatial_scatter` instead.
sc.pl.spatial(